On this page we list some KNIME and TOPPAS workflows which we find useful and worth sharing. Most of them are tested and were successfully used in projects. If you run into trouble executing one of these workflows, please file an issue.
KNIME WORKFLOWS
We are currently building a workflow repository for KNIME so stay tuned. Below you find some workflows we assembled for previous user meetings.
In KNIME, you can import ready-made workflows. To import, click
File -> Import KNIME Workflow…
If you are unfamiliar with the installation process, see our Getting Started page for Workflows.
TARGETED FEATURE DETECTION FOR LABEL-FREE QUANTIFICATION
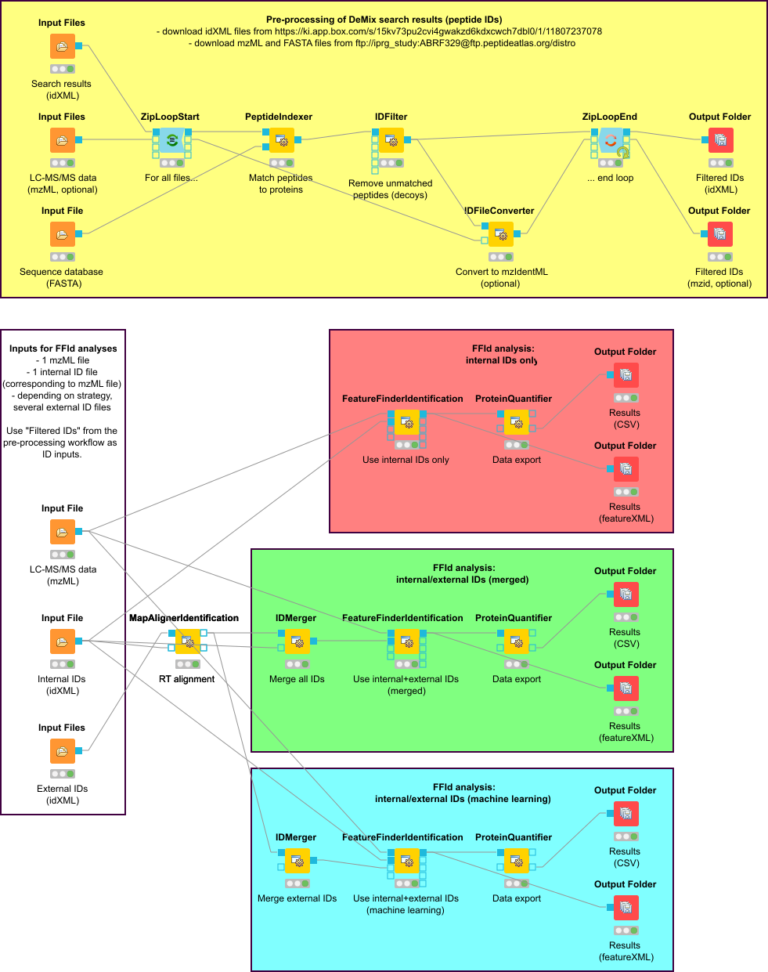
References:
[1] H. Weisser and J. S. Choudhary, “Targeted feature detection for data-dependent shotgun proteomics,” Journal of Proteome Research, 2017.
[Bibtex]
@article{weisser2017targeted,
title={Targeted feature detection for data-dependent shotgun proteomics},
author={Weisser, Hendrik and Choudhary, Jyoti S},
journal={Journal of Proteome Research},
year={2017},
publisher={ACS Publications},
doi={10.1021/acs.jproteome.7b00248}
}
PARALLEL EXECUTION OF OpenMS TOOLS IN KNIME
This workflow shows both the use of standard ZipLoop nodes for iterative OpenMS tool execution, as well as how to execute OpenMS tools in parallel in a high-throughput manner (e.g., to use all computing power for a large number of files)!
DNA HETEROCONJUGATE DETECTION (FLETT ET AL.)
Differential Enzymatic 16O/18O Labelling for the Detection of Cross-Linked Nucleic Acid-Protein Heteroconjugates
NON-TARGETED LC-MS-BASED LIPIDOMICS
This is a lipidomics demonstration workflow as presented during the Metabolomics 2016 conference. It shows how OpenMS can be used in KNIME to cover various steps in MS lipidomics analyses. Used OpenMS algorithms include:
- Detection of lipid features (i.e., m/z, intensity and retention time centroids with their convex peak hulls for a specific lipid ion) in individual samples
- Alignment of samples by retention time
- Finding of corresponding lipid features across samples
- Feature identification via search by accurate mass or spectral matching The workflow further demonstrates how one can use KNIME nodes for interactive visualization, and how to do statistical analyses (exemplified in a discovery section for compounds with significantly differing intensities) with in this case scripting nodes for the statistical language R. The use of KNIME nodes for table operations allows to combine results of various subsections to e.g, annotate quantified compounds of interest with putative identifications, or to find features with probable MS/MS matches and suitable precursor information. If chromatographic information about lipid behaviour is available in sufficient detail, it is also sensible to think about using retention time information for filtering. Our demonstration workflow provides a subsection dedicated to filtering mass-based ID candidates via errors between experimental and predicted retention times. The section in question has previously been shown to achieve very good filtering results for an analysed dataset.
There rarely exists a one-size-fits-all solution pipeline. This is especially true for the still developing field of metabolomics. Here, oftentimes each study requires its own custom-tailored analysis pipeline. In this light, our workflow could be considered more of a starting point or initial template to build upon for your individualized lipidomics analyses. We hope this demonstration workflow could illustrate some of the capabilities of OpenMS in KNIME, and convince you of its modularity and adaptability.
BASIC PEPTIDE IDENTIFICATION
This KNIME workflow shows the very basics of peptide identification using OpenMS.
What it does: It shows how to use a peptide search engine adapter (here: OMSSA) on multiple input files using the same database multiple times.
This workflow is part of the OpenMS tutorial and described in detail in the tutorial.
Preset parameter assumptions: You have recorded MS/MS data in CID mode and medium resolution (comparable to an ion trap)
Important parameters: OMSSAAdapter
- database expects NCBI formatted FASTA files. The .psq filename should be given.
- precursor_mass_tolerance choose according to your acquisition settings
- fragment_mass_tolerance choose according to your acquisition settings
CONSENSUS PEPTIDE IDENTIFICATION
The workflow exemplifies how to increase peptide identification rates in shotgun proteomics experiments by parallel usage of different search engines. The ConsensusID algorithm used is based on the calculation of posterior error probabilities (PEP) and a combination of the normalized scores by considering missing peptide sequences.
What it does: It shows how to apply the ConsensusID algorithm.
This workflow is part of the OpenMS tutorial and described in detail in the tutorial
PEPTIDE IDENTIFICATION AND LABEL-FREE QUANTIFICATION
The workflow exemplifies the analysis capabilities of OpenMS in combination with KNIME. Identification and quantification results are combined and subjected to a simple analysis controlling for the concentration of background peptides being constant and visualize the fold change of investigated peptides across several runs.
What it does: Quantification will be done with the aid of the intensities of so-called features, the peak pattern resulting from peptides (with isotopic representatives) eluting over a certain time. In this example workflow we will use the TOPP tool FeatureFinderCentroided to find those peptide features. Quantifiable features are extracted from the and mapped against the FDR filtered identification results.
This workflow is part of the OpenMS tutorial and described in detail in the tutorial.
Guide for important instrument-specific FeatureFinderCentroided settings:
| Q-TOF | LTQ Orbitrap | |
|---|---|---|
| intensity:bins | 10 | 10 |
| mass_trace:mz_tolerance | 0.02 | 0.004 |
| isotopic_pattern:mz_tolerance | 0.04 | 0.005 |
PROTEIN INFERENCE
This workflow uses a label-free quantification on top of which protein inference and protein quantification are executed.
What it does: Protein inference is performed using FIDO and performs a statistical validation of the protein inference results using advanced KNIME nodes. Protein quantification is done by summarizing all quantified peptides of observed charge states for the inferred proteins.
This workflow is part of the OpenMS tutorial and described in detail in the tutorial.
SWATH ANALYSIS
Example workflow for SWATH analysis using OpenSWATH.
This workflow is part of the OpenMS tutorial and described in detail in the tutorial.
SMALL MOLECULE IDENTIFICATION AND QUANTIFICATION
Example workflow for small molecule identification and quantification.
Features:
- accurate mass search
- detection of differential small molecules
This workflow is part of the OpenMS tutorial and described in detail in the tutorial.
Old (TOPPAS)
PROTEOGENOMICS PEPTIDE IDENTIFICATION
This TOPPAS workflow implements peptide identification and filtering, suitable for finding novel peptides to help improve genome annotations. [1]
Prerequisites: On the software side, the workflow requires OpenMS 2.0.1, the database search engines MS-GF+ and Mascot, as well as the post-processing tools Percolator and Mascot Percolator. On the data side, suitable tandem mass spectrometry data and sequence databases need to be provided.
See the description in the workflow for detailed information about inputs, output and parameters.
References:
[1] H. Weisser, J. C. Wright, J. M. Mudge, P. Gutenbrunner, and J. S. Choudhary, “Flexible Data Analysis Pipeline for High-Confidence Proteogenomics,” Journal of Proteome Research, vol. 15, iss. 12, p. 4686–4695, 2016
[Bibtex]
@article{weisser2016flexible,
title={Flexible Data Analysis Pipeline for High-Confidence Proteogenomics},
author={Weisser, Hendrik and Wright, James C and Mudge, Jonathan M and Gutenbrunner, Petra and Choudhary, Jyoti S},
journal={Journal of Proteome Research},
volume={15},
number={12},
pages={4686--4695},
year={2016},
publisher={ACS Publications},
doi={10.1021/acs.jproteome.6b00765}
}
ITRAQ QUANTITATION AND IDENTIFICATION
This page describes an iTRAQ quantitation and identification workflow.
Assumptions: You have recorded iTRAQ data in HCD/CID mode on an Orbitrap hybrid instrument.
What it does: Peptide quantitation using the ITRAQAnalyzer tool and FDR-controlled identification using three different search engines, which HCD and CID spectra searched separately (for better FDR).
How to run it:
- Install X!Tandem and OMSSA
- Make sure X!Tandem and OMSSA executables are in your PATH, or point the 4 adapter nodes to the executables
- Enter input mzML files in Node #1, and a FASTA database name in Node #8. You also need the preprocessed .phr version of the FASTA file in the same directory (see OMSSAAdapter docu).
- Enter database and server details for both MascotAdapterOnline nodes
- Check the parameter settings in every node, especially ‘IDMapper’